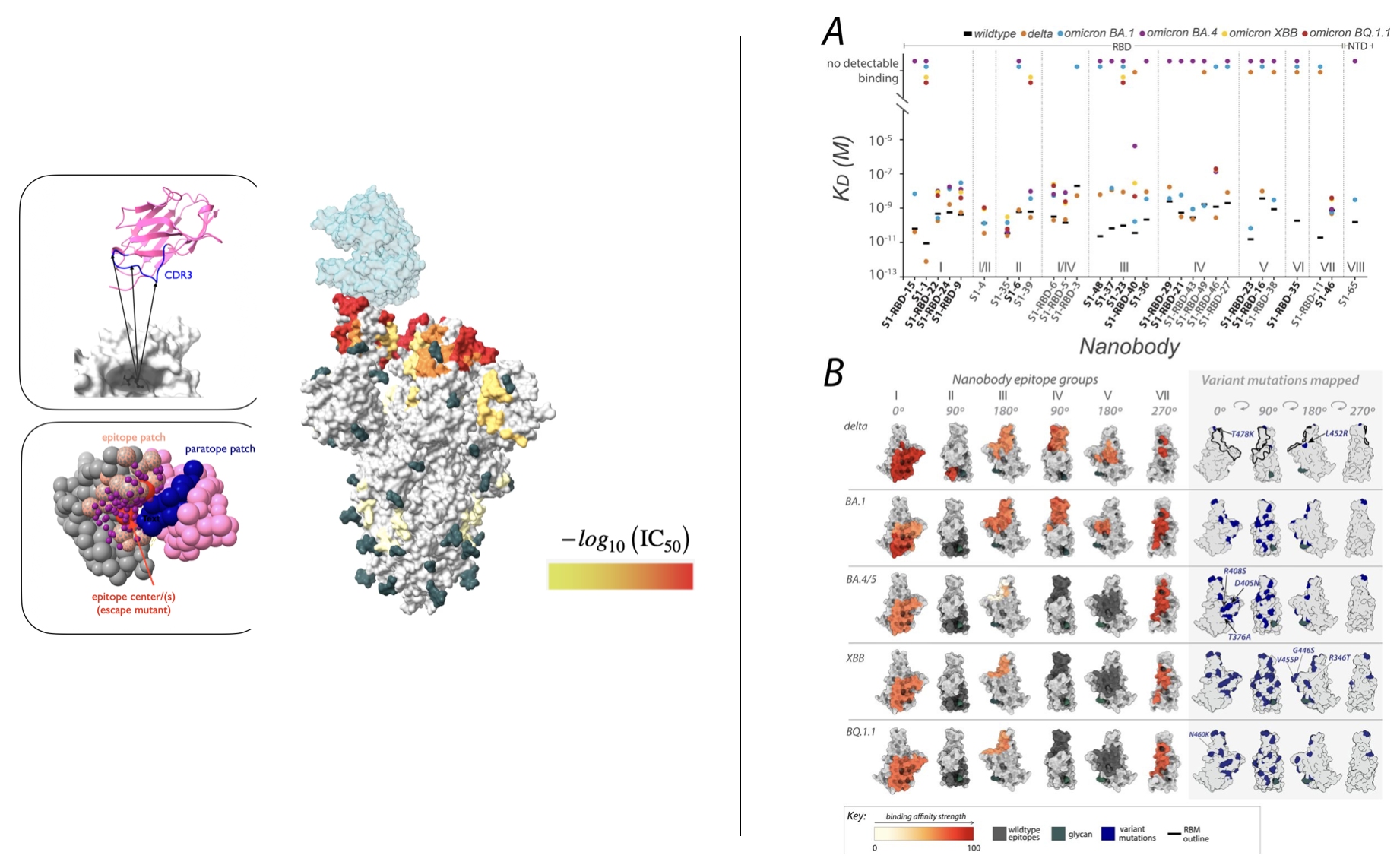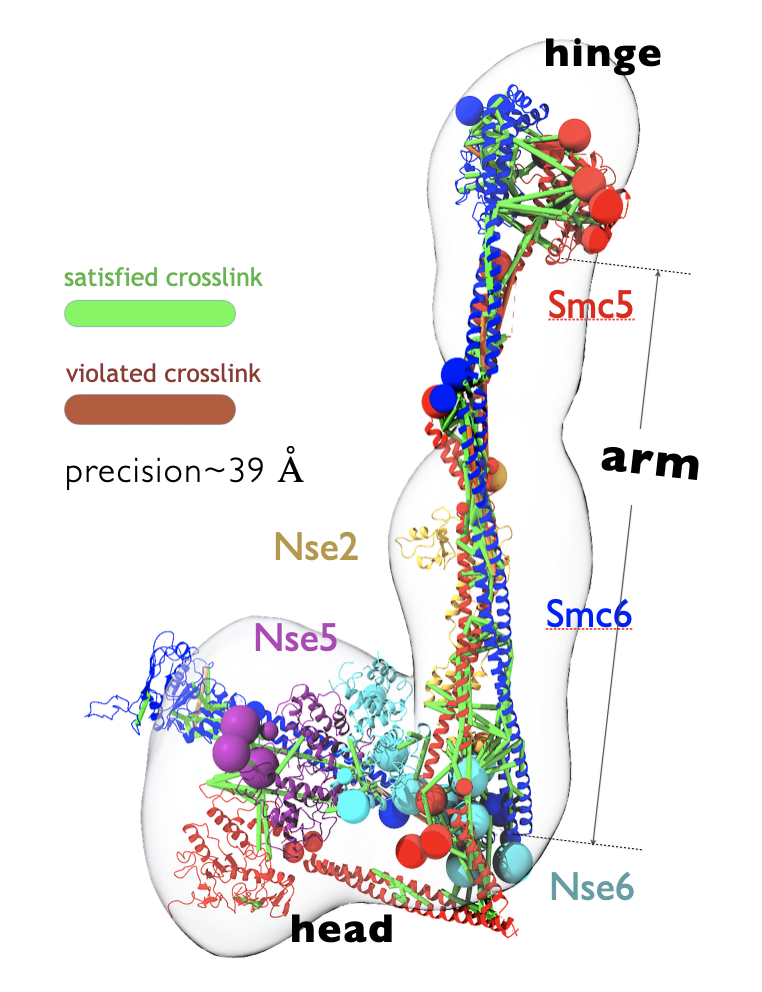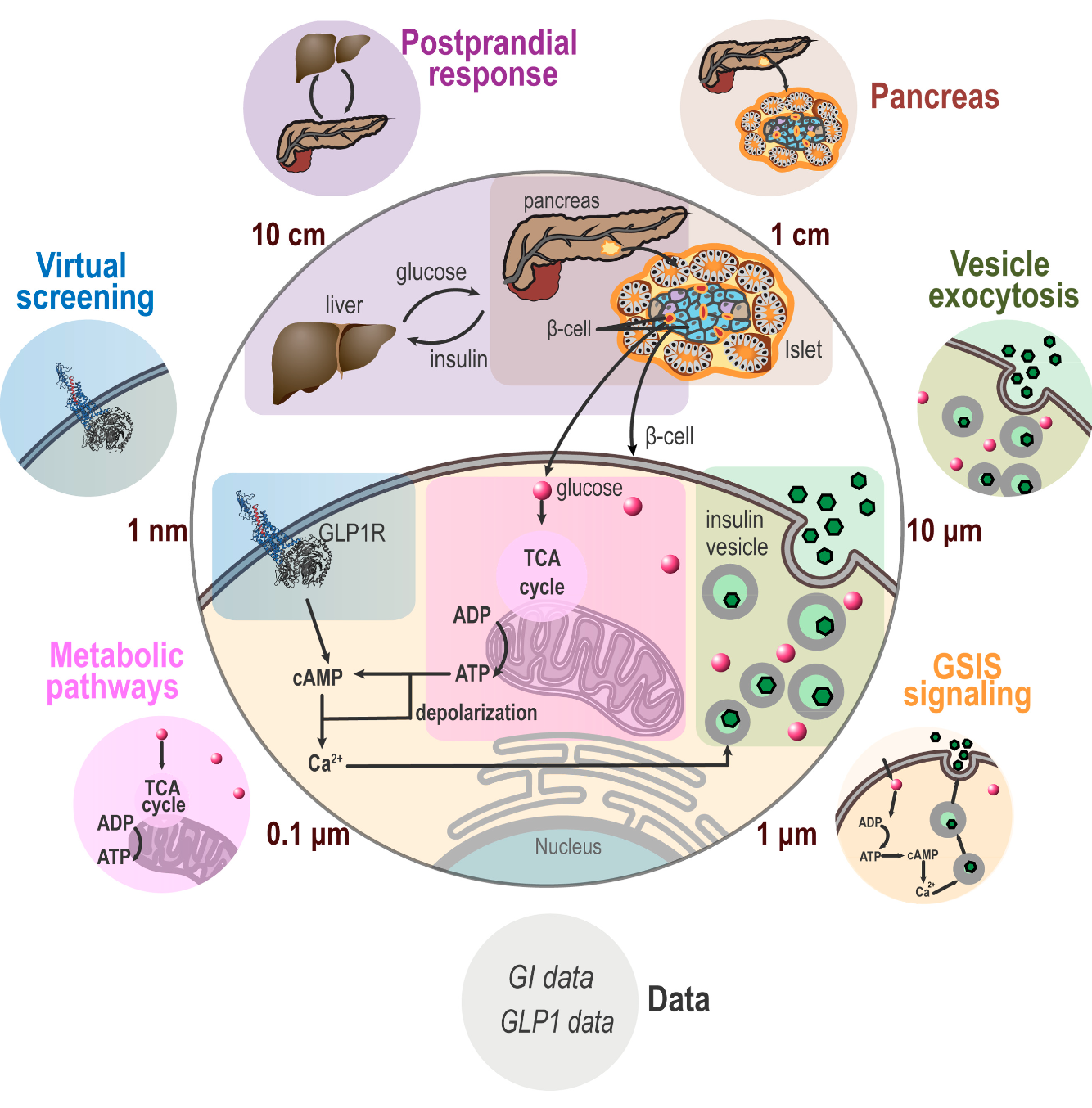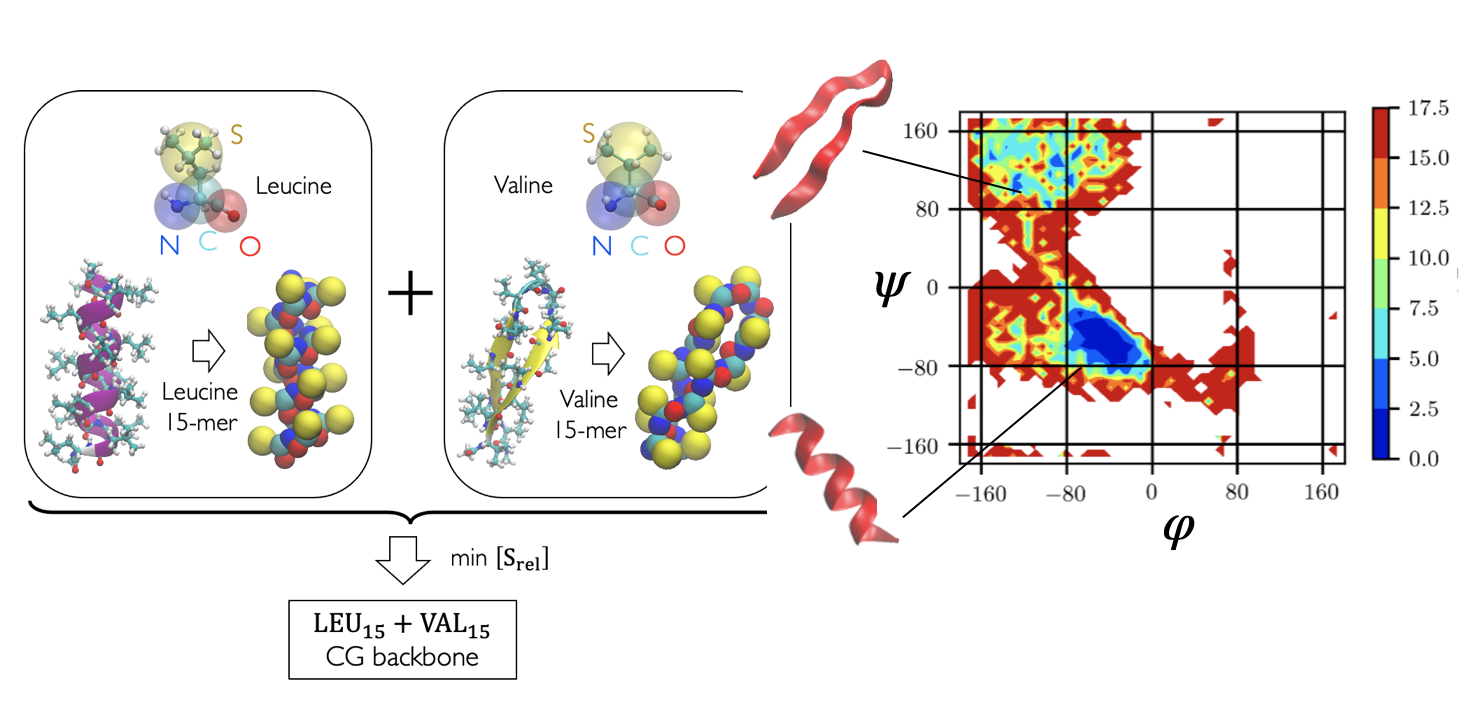
nnprotscan: Scanning PEG-ylation Sites on Peptides for Half-Life Extension
Novo Nordisk
Computational method for systematically scanning chemical modification sites on peptides. Benchmark study on GLP-1 analogs for anti-obesity therapeutics.
peptide designnon-canonical amino acidshalf-lifeopen source